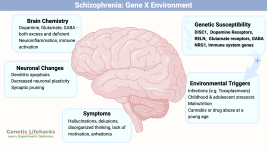
Введение Шизофрения — сложное психическое расстройство, затрагивающее около 1% населения мира. Биологические модели объясняют его через нарушения в генетике, нейротрансмиттерах и структуре мозга. В последние годы (2023-2026) исследования подтвердили полигенную природу заболевания, где сотни генов взаимодействуют с факторами среды. Это отличает шизофрению от моногенных расстройств, подчеркивая роль накопленных мутаций. В статье разберем ключевые механизмы, опираясь на свежие обзоры.
Генетические факторы Генетика играет центральную роль в предрасположенности к шизофреническому спектру. Крупные GWAS-исследования 2025–2026 годов выявили более 100 новых локусов, связанных с риском. Например, гены STAG1 и ZNF136 напрямую ассоциированы с болезнью, влияя на нейронные связи. В российской популяции экзомное секвенирование выделило маркеры вроде HLA-DQB2, связывающие генетику с иммунными нарушениями и когнитивными дефицитами. Исследования близнецов показывают наследуемость до 80%, но эпигенетика (метилирование ДНК) усиливает риск при стрессе или инфекциях. Новые данные из африканских популяций подтверждают общую биологию через глобальные популяции.
Дофаминовая гипотеза: обновления Классическая дофаминовая гипотеза эволюционировала: гиперактивность в нигростриатном пути вызывает позитивные симптомы (галлюцинации), а дефицит в мезокортикальном — негативные (апатия). Актуальные исследования 2025 года подтверждают дисбаланс D2-рецепторов в стриатуме у пациентов с первым психозом. Однако мета-анализ не нашел поддержки для строгой гипотезы в группах высокого риска, предполагая, что дофамин — не единственный фактор. Интеграция с глутаматом: NMDA-антагонисты (как кетамин) вызывают дофаминовый всплеск, имитируя симптомы.
Глутаматная гипотеза Глутаматная гипотеза фокусируется на гипофункции NMDA-рецепторов, приводящей к избыточному глутамату и нейротоксичности. Это объясняет когнитивные дефициты и негативные симптомы лучше дофаминовой модели. Исследования 2020–2026 показывают снижение глутамата в префронтальной коре у пациентов на ранних стадиях. Астроциты регулируют глутамат, и их дисфункция усиливает психоз. Новые данные связывают глутамат с воспалением: повышенные цитокины нарушают баланс, подтверждая нейроиммунную связь.
Нейровизуализация: структурные и функциональные изменения Нейровизуализация раскрывает эпицентры патологии: уменьшение серого вещества в лобных и височных долях, гиперактивность гиппокампа. Мета-анализы 2023-2024 показывают прогрессирующую атрофию в раннем психозе, с эпицентрами в области Брока и фронтоинсулярной коре. fMRI выявляет дисфункцию сетей: сниженная связность в default mode network. PET-исследования SV2A подтверждают снижение синаптической плотности в коре.

(Сравнительный PET-скан мозга при шизофрении.)
Другие биологические аспекты Иммунная дисрегуляция: гены вроде MUC12 и SH3KBP1 связаны с воспалением, а повышенный риск ассоциирован с истончением сетчатки (маркер нейровоспаления). Нейроразвитие: нарушения в подростковом периоде, включая синаптический pruning, усугубляют риск.
Оценка и критика биологических моделей Биологические подходы детерминистичны, игнорируя среду (стресс, инфекции). Редукционизм упрощает: шизофрения - биопсихосоциальное расстройство. Однако интеграция моделей (дофамин + глутамат) перспективна.
Лечение на основе биологии Антипсихотики (типичные/атипичные) таргетируют дофамин, но новые препараты вроде Cobenfy (2024) влияют на мускариновые рецепторы, снижая побочки. Глутаматные модуляторы (rolipram) в испытаниях. Персонализированная медицина: генетические тесты для подбора терапии.
Заключение Биологические объяснения шизофрении эволюционировали от моногипотез к интегративным моделям. Будущие исследования фокусируются на мультиомиксе и AI для предикции. Это открывает путь к превентивным стратегиям.
Часто задаваемые вопросы (FAQ)
Генетические факторы Генетика играет центральную роль в предрасположенности к шизофреническому спектру. Крупные GWAS-исследования 2025–2026 годов выявили более 100 новых локусов, связанных с риском. Например, гены STAG1 и ZNF136 напрямую ассоциированы с болезнью, влияя на нейронные связи. В российской популяции экзомное секвенирование выделило маркеры вроде HLA-DQB2, связывающие генетику с иммунными нарушениями и когнитивными дефицитами. Исследования близнецов показывают наследуемость до 80%, но эпигенетика (метилирование ДНК) усиливает риск при стрессе или инфекциях. Новые данные из африканских популяций подтверждают общую биологию через глобальные популяции.
Дофаминовая гипотеза: обновления Классическая дофаминовая гипотеза эволюционировала: гиперактивность в нигростриатном пути вызывает позитивные симптомы (галлюцинации), а дефицит в мезокортикальном — негативные (апатия). Актуальные исследования 2025 года подтверждают дисбаланс D2-рецепторов в стриатуме у пациентов с первым психозом. Однако мета-анализ не нашел поддержки для строгой гипотезы в группах высокого риска, предполагая, что дофамин — не единственный фактор. Интеграция с глутаматом: NMDA-антагонисты (как кетамин) вызывают дофаминовый всплеск, имитируя симптомы.
Глутаматная гипотеза Глутаматная гипотеза фокусируется на гипофункции NMDA-рецепторов, приводящей к избыточному глутамату и нейротоксичности. Это объясняет когнитивные дефициты и негативные симптомы лучше дофаминовой модели. Исследования 2020–2026 показывают снижение глутамата в префронтальной коре у пациентов на ранних стадиях. Астроциты регулируют глутамат, и их дисфункция усиливает психоз. Новые данные связывают глутамат с воспалением: повышенные цитокины нарушают баланс, подтверждая нейроиммунную связь.
Нейровизуализация: структурные и функциональные изменения Нейровизуализация раскрывает эпицентры патологии: уменьшение серого вещества в лобных и височных долях, гиперактивность гиппокампа. Мета-анализы 2023-2024 показывают прогрессирующую атрофию в раннем психозе, с эпицентрами в области Брока и фронтоинсулярной коре. fMRI выявляет дисфункцию сетей: сниженная связность в default mode network. PET-исследования SV2A подтверждают снижение синаптической плотности в коре.
(Сравнительный PET-скан мозга при шизофрении.)
Другие биологические аспекты Иммунная дисрегуляция: гены вроде MUC12 и SH3KBP1 связаны с воспалением, а повышенный риск ассоциирован с истончением сетчатки (маркер нейровоспаления). Нейроразвитие: нарушения в подростковом периоде, включая синаптический pruning, усугубляют риск.
Оценка и критика биологических моделей Биологические подходы детерминистичны, игнорируя среду (стресс, инфекции). Редукционизм упрощает: шизофрения - биопсихосоциальное расстройство. Однако интеграция моделей (дофамин + глутамат) перспективна.
Лечение на основе биологии Антипсихотики (типичные/атипичные) таргетируют дофамин, но новые препараты вроде Cobenfy (2024) влияют на мускариновые рецепторы, снижая побочки. Глутаматные модуляторы (rolipram) в испытаниях. Персонализированная медицина: генетические тесты для подбора терапии.
Заключение Биологические объяснения шизофрении эволюционировали от моногипотез к интегративным моделям. Будущие исследования фокусируются на мультиомиксе и AI для предикции. Это открывает путь к превентивным стратегиям.
Часто задаваемые вопросы (FAQ)
- Каковы биологические причины шизофрении? Генетика (полигенные риски), дисбаланс дофамина/глутамата, нейронные изменения.
- Шизофрения биологическая или психологическая? Комбинация: биология создает уязвимость, психология - триггеры.
- Почему избыток дофамина вызывает психоз? Активирует D2-рецепторы, вызывая галлюцинации.
- Три биологических фактора риска? Генетика, нейронные различия, дофаминовый дисбаланс.